

PCR (polymerase chain reaction)
Let's say you have a biological sample with trace amounts of DNA in it. You want to work with the DNA, perhaps characterize it by sequencing, but there isn't much to work with. This is where PCR comes in. PCR is the amplification of a small amount of DNA into a larger amount. It is quick, easy, and automated. Larger amounts of DNA mean more accurate and reliable results for your later techniques.
PCR can be used to create a DNA "fingerprint," which is unique to each individual. These DNA fingerprints can be useful in real-world applications relating to paternity/maternity, kinship, and forensic testing.
 The technique was developed by Nobel laureate biochemist Kary Mullis in 1984 and is based on the discovery of the biological activity at high temperatures of DNA polymerases found in thermophiles (bacteria that live in hot springs).
The technique was developed by Nobel laureate biochemist Kary Mullis in 1984 and is based on the discovery of the biological activity at high temperatures of DNA polymerases found in thermophiles (bacteria that live in hot springs).
Most DNA polymerases (enzymes that make new DNA) work only at low temperatures. But at low temperatures DNA is tightly coiled, so the polymerases don't stand much of a chance of getting at most parts of the molecules.
But these thermophile DNA polymerases work at 100C, a temperature at which DNA is denatured (in linear form). This thermophilic DNA polymerase is called Taq polymerase, named after Thermus aquaticus, the bacteria it is derived from.
Taq polymerase, however, has no proofreading ability. Other thermally stable polymerases, such as Vent and Pfu, have been discovered to both work for PCR and to proofread.
You'll need four things to perform PCR on a sample:
1. The target sample. This is the biological sample you want to amplify DNA from.
2. A primer. Short strands of DNA that adhere to the target segment. They identify the portion of DNA to be multiplied and provide a starting place for replication.
3. Taq polymerase. This is the enzyme that is in charge of replicating DNA. This is the polymerase part of the name polymerase chain reaction.
4. Nucleotides. You'll need to add nucleotides (dNTPs) so the DNA polymerase has building blocks to work with.
There are three major steps to PCR and they are repeated over and over again, usually 25 to 75 times. This is where the automation is most appreciated.
1. Your target sample is heated. This denatures the DNA, unwinding it and breaking the bonds that hold together the two strands of the DNA molecule, leaving you with single stranded DNA (ssDNA).
2. Temperature is reduced and the primer is added. The primer molecules now have the opportunity to bind (anneal) to the pieces of ssDNA. This labels the portions of DNA to be amplified and provides a starting place for replication.
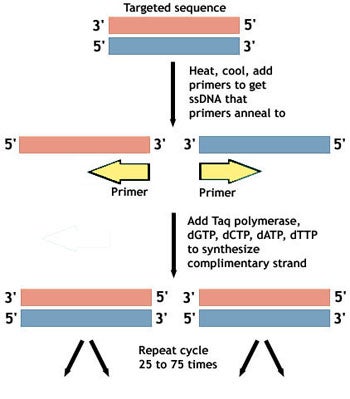 3. New pieces of ssDNA are made. Taq polymerase catalyzes the generation of new pieces of ssDNA that are complimentary to the portions marked by the primers. The job of Taq polymerase is to move along the strand of DNA and use it as a template for assembling a new strand that is complimentary to the template. This is the chain reaction in the name polymerase chain reaction.
3. New pieces of ssDNA are made. Taq polymerase catalyzes the generation of new pieces of ssDNA that are complimentary to the portions marked by the primers. The job of Taq polymerase is to move along the strand of DNA and use it as a template for assembling a new strand that is complimentary to the template. This is the chain reaction in the name polymerase chain reaction.
PCR is so efficient because it multiplies the DNA exponentially for each of the 25 to 75 cycles. A cycle takes only a minute or so and each new segment of DNA that is made can serve as a template for new ones.
Perhaps the most important thing to remember is to be very aware of contamination. If, for example, you unknowingly slough off a piece of skin into your sample, then your DNA may be amplified in the PCR reaction. Here are some other factors to optimize your results with PCR:
1. Annealing temperature. Starts at the low end of what you think will work, then move up as necessary. If the temperature is too low, the primers will make more mistakes and you'll get too many bands when you run your sample on a gel. If the temperature is too high you will get no results and your gel will be blank. You want to be about 3C to 5C below the melting temperature (Tm). A rough formula for determining Tm is Tm=4(G + C) + 2(A + T).
2. Magnesium concentration. You want your Mg2+ concentration to be about 1.5mM to 3mM. If you go too high, the polymerase will make more mistakes.
3. Think carefully about primer design. Both primers should have approximately the same Tm so they both anneal at the same temperature. Two out of three bases on the 3' end should be G or C to get good hybridization (G and C have three H-bonds so you get better polymerization). Lastly, avoid primer dimers, which occur when the primers have ends that will anneal to each other. This will produce NO product.
4. More is not necessarily better. More polymerase produces more nonspecific product, so don't just carelessly dump in a bunch of polymerase. Additionally, PCR reactions don't work if there is too much DNA.
RT-PCR
Taq polymerase does not work on RNA samples, so PCR cannot be used to directly amplify RNA molecules. The incorporation of the enzyme reverse transcriptase (RT), however, can be combined with traditional PCR to allow for the amplification of RNA molecules. After you add your RNA sample to the PCR machine, add a DNA primer as usual and allow it to anneal to your target molecule. Then add RT along with dNTPs, which will elongate the DNA primer and make a cDNA copy of the RNA molecules and run the PRC reaction as usual. The product of RT-PCR is a double stranded DNA molecule analogous to the target segment of the RNA molecule.
Read more about: Mama Ji's Molecular Kitchen
You may need to edit author's name to meet the style formats, which are in most cases "Last name, First name."
https://askabiologist.asu.edu/pcr-polymerase-chain-reaction
Bibliographic details:
- Article: PCR (polymerase chain reaction)
- Author(s): Dr. Biology
- Publisher: Arizona State University School of Life Sciences Ask A Biologist
- Site name: ASU - Ask A Biologist
- Date published:
- Date accessed:
- Link: https://askabiologist.asu.edu/pcr-polymerase-chain-reaction
APA Style
Dr. Biology. (). PCR (polymerase chain reaction). ASU - Ask A Biologist. Retrieved from https://askabiologist.asu.edu/pcr-polymerase-chain-reaction
American Psychological Association. For more info, see http://owl.english.purdue.edu/owl/resource/560/10/
Chicago Manual of Style
Dr. Biology. "PCR (polymerase chain reaction)". ASU - Ask A Biologist. . https://askabiologist.asu.edu/pcr-polymerase-chain-reaction
Dr. Biology. "PCR (polymerase chain reaction)". ASU - Ask A Biologist. . ASU - Ask A Biologist, Web. https://askabiologist.asu.edu/pcr-polymerase-chain-reaction
For more info, see http://owl.english.purdue.edu/owl/resource/717/04/
MLA 2017 Style
Modern Language Association, 7th Ed. For more info, see http://owl.english.purdue.edu/owl/resource/747/08/

Be Part of
Ask A Biologist
By volunteering, or simply sending us feedback on the site. Scientists, teachers, writers, illustrators, and translators are all important to the program. If you are interested in helping with the website we have a Volunteers page to get the process started.